Scooby Evaluator¶
General Imports¶
[1]:
%load_ext autoreload
%autoreload 2
[2]:
import os
import numpy as np
import pandas as pd
import scipy
import torch
import tqdm
from accelerate import Accelerator
from peft import LoraConfig, get_peft_model
from torch.utils.data import DataLoader
from enformer_pytorch.data import GenomeIntervalDataset
from scooby.modeling import Scooby
from scooby.data import onTheFlyDataset, onTheFlyPseudobulkDataset
from scooby.utils.utils import fix_rev_comp_multiome, undo_squashed_scale, get_pseudobulk_count_pred,get_gene_slice_and_strand
[3]:
data_path = '/data/ceph/hdd/project/node_08/QNA/scborzoi/neurips_bone_marrow/'
scooby¶
Functions for Pearson correlation¶
[4]:
def stack_and_pearson(x,y):
return torch.corrcoef(torch.stack([x,y]))[0,1]
batched_pearson = torch.vmap(stack_and_pearson, chunk_size=1)
def predict(model, seqs, seqs_rev_comp, conv_weights, conv_biases, bins_to_predict = None):
bs = seqs.shape[0]
# assert bs == 1
with torch.no_grad():
with torch.autocast("cuda"):
outputs = model.forward_sequence_w_convs(seqs, conv_weights, conv_biases, bins_to_predict = bins_to_predict)
if bins_to_predict is not None:
outputs_rev_comp = model.forward_sequence_w_convs(seqs_rev_comp, conv_weights, conv_biases, bins_to_predict = (6143 - bins_to_predict))
else:
outputs_rev_comp = model.forward_sequence_w_convs(seqs_rev_comp, conv_weights, conv_biases, bins_to_predict = None)
flipped_version = torch.flip(outputs_rev_comp,(1,-3))
outputs_rev_comp_test = fix_rev_comp_multiome(flipped_version) #fix_rev_comp2(flipped_version)
return (outputs + outputs_rev_comp_test)/2
Load the model¶
[5]:
accelerator = Accelerator(step_scheduler_with_optimizer = False)
Detected kernel version 4.18.0, which is below the recommended minimum of 5.5.0; this can cause the process to hang. It is recommended to upgrade the kernel to the minimum version or higher.
Option 1: Load pretrained scooby from the hub¶
[6]:
csb = Scooby.from_pretrained(
'johahi/neurips-scooby',
cell_emb_dim=14,
embedding_dim=1920,
n_tracks=3,
return_center_bins_only=True,
disable_cache=False,
use_transform_borzoi_emb=True,
)
[7]:
clip_soft = 5
[8]:
csb = accelerator.prepare(csb)
[9]:
context_length = 524288
Option 2: Load self-trained scooby checkpoint, un-lora it, and save it¶
[ ]:
import safetensors
from scooby.utils.utils import get_lora
#Initialize the config
csb = Scooby.from_pretrained(
'johahi/borzoi-replicate-0',
cell_emb_dim=14,
embedding_dim=1920,
n_tracks=3,
return_center_bins_only=True,
disable_cache=False,
use_transform_borzoi_emb=True,
)
# we have to make the lora version, load the weights, and merge the lora weights back into scooby
csb = get_lora(csb, train=False)
print (safetensors.torch.load_model(csb,'<<checkpoint_path>>/model.safetensors'))
csb = csb.merge_and_unload()
# If you want to save the model to load it quicker next time
csb.save_pretrained('test_scooby_checkpoint')
# can be then loaded with
# scooby = Scooby.from_pretrained('test_scooby_checkpoint', cell...)
Specify genome¶
[10]:
gtf_file = os.path.join(data_path, "gencode.v32.annotation.sorted.gtf.gz")
fasta_file = os.path.join(data_path, 'genome_human.fa')
bed_file = os.path.join(data_path, 'sequences.bed')
Load neighbors and embedding¶
[11]:
base_path = os.path.join(data_path, 'scooby_training_data', 'pseudobulks/')
[12]:
sample = 'merged'
neighbors = scipy.sparse.load_npz(os.path.join(data_path, 'scooby_training_data', 'no_neighbors.npz'))
embedding = pd.read_parquet(os.path.join(data_path, 'scooby_training_data', 'embedding_no_val_genes_new.pq'))
[13]:
cell_type_index = pd.read_parquet(os.path.join(data_path, 'scooby_training_data/celltype_fixed.pq'))
cell_type_index['size'] = cell_type_index['cellindex'].apply(lambda x: len(x))
[14]:
cell_type_index['celltype'] = cell_type_index['celltype'].str.replace(' ', '_').replace(r"G/M_prog", "G+M_prog").replace("MK/E_prog", "MK+E_prog") #+ '_filtered'
cell_type_index = cell_type_index.sort_values('celltype')
Eval on Val¶
Sequence dataloader¶
This only loads the sequences in the right order
[15]:
gtf_file = f"{data_path}/gencode.v32.annotation.gtf"
[16]:
from scooby.utils.transcriptome import Transcriptome
[17]:
transcriptome = Transcriptome(gtf_file)
[18]:
import polars as pl
filter_val = lambda df: df.filter(True)#
val_ds = GenomeIntervalDataset(
bed_file = os.path.join(data_path,'scooby_training_data_fixed', 'test_gene_sequences.csv'),
fasta_file = fasta_file,
filter_df_fn = lambda df: df.filter((pl.col('column_2') >=0)),
return_seq_indices = False,
shift_augs = (0,0),
rc_aug = False,
return_augs = True,
context_length = context_length,
chr_bed_to_fasta_map = {}
)
len(val_ds)
[18]:
2550
[19]:
val_dataset = onTheFlyDataset(
None,
None,
embedding,
val_ds,
neighbors=neighbors,
get_targets= False,
random_cells = False,
cells_to_run = None,
clip_soft = clip_soft,
)
val_loader = DataLoader(val_dataset, batch_size=1, shuffle = False, num_workers = 1)
csb, val_loader = accelerator.prepare(csb, val_loader)
clip_soft
[19]:
5
Target dataloader on bigwig pseudobulk files¶
[20]:
val_dataset_targets = onTheFlyPseudobulkDataset(
cell_types = cell_type_index['celltype'].values,
ds = val_ds,
base_path = base_path,
)
Get cell conv_weights for all cells¶
[21]:
csb.eval()
cell_indices = []
size_factors_per_ct = []
for _, row in tqdm.tqdm(cell_type_index.iterrows(),disable = True):
cell_indices.append(
torch.from_numpy(
np.vstack(
embedding.iloc[row['cellindex']]['embedding'].values # gets embeddings of all cells of the cell type
)
).unsqueeze(0)
) # prep cell_embeddings
# get conv weights and biases for all cells sorted by cell type in a list
cell_emb_conv_weights_and_biases = []
for cell_emb_idx in tqdm.tqdm(cell_indices, disable = True):
cell_emb_idx = cell_emb_idx.cuda()
conv_weights, conv_biases = csb.forward_cell_embs_only(cell_emb_idx)
cell_emb_conv_weights_and_biases.append((conv_weights.to(torch.float16), conv_biases.to(torch.float16)))
Get counts over exons¶
[22]:
num_neighbors = 1
all_outputs, all_targets = [], []
val_dataset_target_loader = iter(DataLoader(val_dataset_targets, batch_size=1, shuffle = False, num_workers = 4))
# iterate over all gene sequences
for i,x in tqdm.tqdm(enumerate(val_loader), disable = False, total=len(val_dataset)):
gene_slice, strand = get_gene_slice_and_strand(transcriptome, val_dataset_targets.genome_ds.df[i, 'column_4'], val_dataset_targets.genome_ds.df[i, 'column_2'], span = False)
# continue
targets = (next(val_dataset_target_loader)[2]).float().cuda()
if len(gene_slice) == 0:
continue
bs = x[0].shape[0]
seqs = x[0].cuda().permute(0,2,1)
stacked_outputs = []
stacked_outputs = get_pseudobulk_count_pred(
csb = csb,
seqs = seqs,
cell_emb_conv_weights_and_biases = cell_emb_conv_weights_and_biases,
gene_slice = gene_slice,
strand = strand,
model_type = "multiome",
predict = predict,
clip_soft = clip_soft,
num_neighbors = 1
)
temp_stack = []
all_outputs.append(stacked_outputs)
if strand == '+':
all_targets.append(undo_squashed_scale(targets[0, gene_slice, ::2], clip_soft=384).sum(axis=0).detach().clone().cpu().squeeze())
elif strand == '-':
all_targets.append(undo_squashed_scale(targets[0, gene_slice, 1::2], clip_soft=384).sum(axis=0).detach().clone().cpu().squeeze())
all_outputs, all_targets = torch.vstack(all_outputs).clone().numpy(force=True),torch.vstack(all_targets).clone().numpy(force=True)
100%|██████████| 2550/2550 [17:32<00:00, 2.42it/s]
[23]:
import matplotlib.pyplot as plt
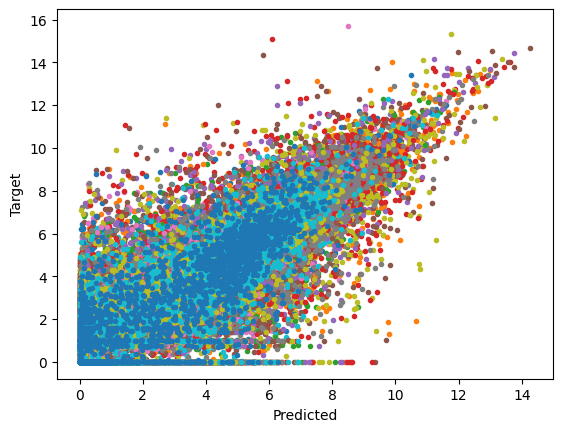
plt.plot(np.log2((all_outputs+1)), np.log2(all_targets+1), '.')
plt.xlabel('Predicted')
plt.ylabel('Target')
[23]:
Text(0, 0.5, 'Target')
[24]:
spearmanr = [
scipy.stats.spearmanr(np.log2(all_outputs[:, i] + 1),
np.log2(all_targets[:, i] + 1))[0] for i in range(all_outputs.shape[1])
]
pearsonr = [
scipy.stats.pearsonr (np.log2(all_outputs[:, i] + 1),
np.log2(all_targets[:, i] + 1))[0] for i in range(all_outputs.shape[1])]
[25]:
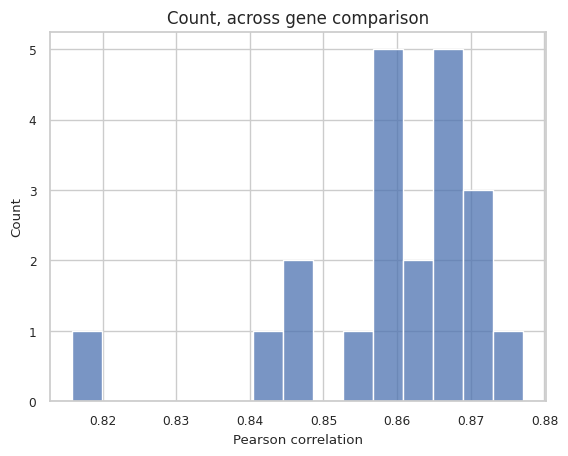
np.mean(pearsonr), np.median(pearsonr) # np.mean(pearsonr), np.median(pearsonr) with normos (0.8695861071966647, 0.8775566542956221)
[25]:
(np.float32(0.8594968), np.float32(0.8618465))
[26]:
import seaborn as sns
sns.set_theme(style="whitegrid", font_scale=0.8)
sns.histplot(pearsonr, bins = 15)
plt.xlabel('Pearson correlation')
plt.title('Count, across gene comparison', size = 12)
[26]:
Text(0.5, 1.0, 'Count, across gene comparison')
[27]:
# log2(x+1)
counts_predicted_log = np.log2(pd.DataFrame(all_outputs) + 1) #pd.read_parquet("/data/ceph/hdd/project/node_08/QNA/scborzoi/neurips_bone_marrow/eval/count_predicted_log.parquet")# np.log2(pd.DataFrame(all_outputs) + 1)
#counts_predicted_log = counts_predicted_log.drop(16, axis = 1) #remove other myloids?
# substract mean per gene across all cell types
counts_predicted_log = counts_predicted_log - counts_predicted_log.mean(axis=0)
#substract mean per cell type across all genes
counts_predicted_log = counts_predicted_log.subtract(counts_predicted_log.mean(axis=1), axis=0)
counts_target_log =np.log2(pd.DataFrame(all_targets) + 1)# pd.read_parquet("/data/ceph/hdd/project/node_08/QNA/scborzoi/neurips_bone_marrow/eval/count_target_log.parquet")# np.log2(pd.DataFrame(all_targets) + 1)
#counts_target_log = counts_target_log.drop(16, axis = 1) #remove other myloids?
counts_target_log = counts_target_log - counts_target_log.mean(axis=0)
counts_target_log = counts_target_log.subtract(counts_target_log.mean(axis=1), axis=0)
[28]:
import matplotlib as mp
font_size= 12
mp.rcParams['axes.labelsize'] = font_size
mp.rcParams['axes.linewidth'] = font_size / 24.
mp.rcParams['axes.titlesize'] = font_size
mp.rcParams['legend.fontsize'] = font_size
mp.rcParams['xtick.labelsize'] = font_size
mp.rcParams['ytick.labelsize'] = font_size
[29]:
from matplotlib import pyplot as plt
from matplotlib.colors import LogNorm
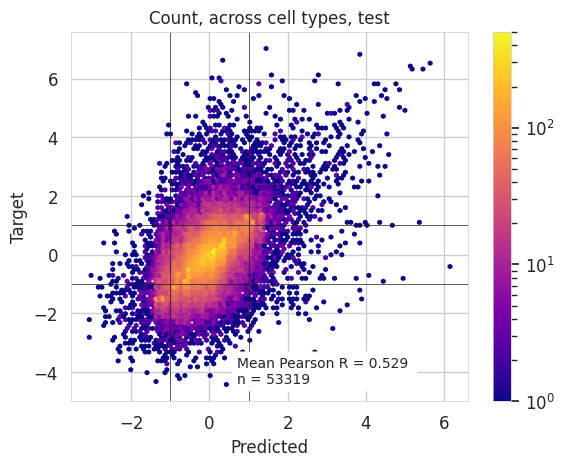
ax = plt.hexbin(x=counts_predicted_log.values.ravel(), y=counts_target_log.values.ravel(), mincnt=1, cmap='plasma', norm=LogNorm())
plt.axvline(1, c='k', linewidth = font_size/24)
plt.axvline(-1,c='k', linewidth = font_size/24)
plt.axhline(1,c='k', linewidth = font_size/24)
plt.axhline(-1,c='k', linewidth = font_size/24)
plt.colorbar()
plt.ylabel('Target')
plt.title('Count, across cell types, test')
p=scipy.stats.pearsonr(counts_predicted_log.values.ravel(), counts_target_log.values.ravel())[0]
# plt.text(2.5,-5, f"Pearson corr.: {np.round(p, 3)}", bbox=dict(facecolor='white', edgecolor='black', boxstyle='round'))
plt.text(.7,-4.4, f"Mean " + r"Pearson R"+ f" = {p:.3f} \nn = {len(counts_predicted_log.values.ravel())}", bbox=dict(facecolor='white', edgecolor='white', boxstyle='round'), fontdict={'size' : 10})
print(p)
plt.xlabel('Predicted')
#plt.savefig("Fig2E.svg",bbox_inches="tight")
0.5291457
[29]:
Text(0.5, 0, 'Predicted')
[ ]: